OVERVIEW of BIONEER QA/QC
1. Introduction
2. Custom Analytical Service
3. Custom Analytical Service for Antisense Oligonucleotide
4. MALDI-TOF QC System
5. HPLC
6. GC
7. NMR
8. Test of Heavy Metal Content
9. Test of Water Content
10. Bioburden test
11. Endotoxin test
Introduction
At Bioneer, quality control is fundamental to our manufacturing processes and guarantees high quality product.
Bioneer's Quality Control for Oligonucleotides can be divided into three distinct areas:
1. Bioneer's Olignucleotide Ordering Systems:
Orders from customers are gathered on a main production server system prior to synthesis. To eliminate re-entry errors,
on-line and e-mail orders are recommended. Orders are automatically distributed (batched) to an appropriate synthesizer
according to the length of oligo, the type of modification, and users’ plate choice. Every lot to be synthesized is
labeled with its own Barcode ID, which is used for identifying the oligonucleotide plate through the synthesis process.
Bioneer's Quality Assurance Staff can monitor all procedures from synthesis to aliquoting using our proprietary
Automatic Oligonucleotide Production System (AOP System).
2. Automatic MALDI-TOF QC:
Bioneer employs multiple MALDI-TOF mass spectrometers that are fully automated from loading to mass determination.
The mass spectrometry data for each sample is automatically inserted into the oligo information sheet. Bioneer is one
of the few oligo producers that checks all oligonucleotides (single and high throughput orders) by MALDI-TOF and
provides mass data with each oligo, free of charge.

Figure 1. Typical Oligo Datasheet with MALDI-TOF Information
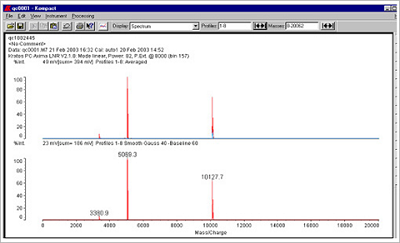
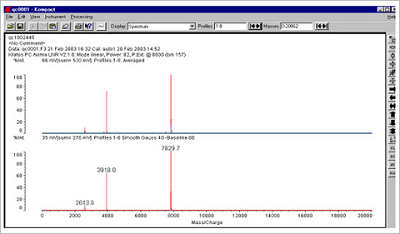
Figure 2. Examples of a typical 30 mer and 23 mer oligo spectrum, in this case employing a Kratos MALDI-TOF system.
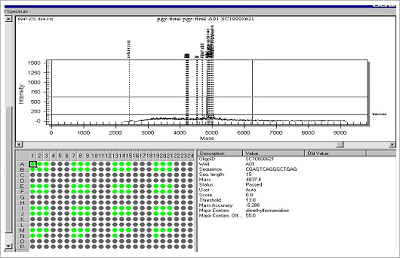
Figure 3. Example of High Throughput Plate QC Analysis.
The mass spectrum and result of 92 (plate) oligonucleotides – Bioneer can also provide “spectrochecked” oligo QC data for users of the Sequenom SNP Analysis System.
3. QC of Large Scale, Antisense and Decoy Oligonucleotides:
The high quality oligonucleotides used in gene therapy and other therapeutic-type applications obviously require
more stringent QC. Bioneer has developed specialized QC methods for large scale, antisense and decoy oligonucleotides.
These specialized QC procedures comprise HPLC, CE, gel electrophoresis, NMR and additional specific tests such as
endotoxin, bioburden, moisture content and etc. Bioneer's wide array of Q.C. procedures satisfies all of our users'
specific QC requirements. The array of QC steps available from Bioneer is as follows:
1. Ion-Exchange HPLC analysis
2. Reverse Phase HPLC analysis
3. Capillary Electrophoresis
4. NMR analysis
5. Moisture content analysis
6. Sodium content analysis
7. Heavy metal content analysis
8. Solvent content analysis
9. Endotoxin test
10. Bioburdent test
Bioneer can also supply reports of API stability testing under the contract period. For information on pricing and
ordering, as well as additional information on any of Bioneer's specialized QC procedures, please e-mail
order.usa@bioneer.us.com .
Custom Analytical Service
| I-E HPLC |
25 |
25 |
25 |
25 |
50 |
50 |
| RP HPLC |
20 |
20 |
20 |
20 |
40 |
40 |
| Capillary electrophoresis |
30 |
30 |
30 |
30 |
30 |
30 |
| MALDI-TOF analysis, Spectrocheck*1 |
1 |
1 |
1 |
1 |
2 |
2 |
| MALDI-TOF analysis, other mass system |
Free |
Free |
Free |
Free |
2 |
2 |
*1 : For those users who ordered more than 96 oligonucleotides, QC will be $100 for each plate.
If the oligonucleotides were ordered less than 96 oligonucleotides in separate tubes, please refer to the
price above in the table. Data will be provided on CD or paper; however, if the oligonucleotides are more than 96,
it will be provided in CD format.
Custom Analytical Service for Antisense Oligonucleotide
| I-E HPLC |
25 |
25 |
25 |
25 |
50 |
50 |
| RP HPLC |
20 |
20 |
20 |
20 |
40 |
40 |
| Capillary electrophoresis |
30 |
30 |
30 |
30 |
30 |
30 |
| NMR analysis |
|
|
|
|
20 |
20 |
| PAGE analysis |
15 |
15 |
15 |
15 |
15 |
15 |
| Moisture content analysis |
|
|
|
|
35 |
35 |
| Metal content analysis |
|
|
|
|
50 |
50 |
| Solvent content analysis |
|
|
|
|
35 |
35 |
| Endotoxin test(LAL) |
|
|
|
|
50 |
50 |
| Bioburden test |
|
|
|
|
100 |
100 |
| MALDI-TOF analysis, other mass system |
2 |
2 |
2 |
2 |
2 |
2 |
MALDI-TOF QC System
The Use of Matrix-Assisted Laser Desorption / Ionization Time of Flight Mass Spectrometry of Synthetic
Oligonucleotide QC
At Bioneer MALDI-TOF (Matrix Assisted Laser Desorption-Ionization Time of Flight) is the technology used extensively
for failure detection and other problems that cannot be resolved by other methods. Bioneer's fully automated,
high throughput QC systems allow the company to provide superior, high quality product superior to that of our
competitors. The QC systems installed at Bioneer currently can check the quality of 35,000 synthetic oligonucleotides
per day. Each and every oligo is supplied with an oligo data sheet that includes MALDI-TOF mass spectrum.
Interpreting MALDI-TOF Mass QC for Oligonucleotide
A MALDI-TOF mass spectrometer accurately measures molecular weight of a sample. The technique is most useful because
it compares the theoretical mass calculated on the basis of oligonucleotide sequence to actual measured data. MALDI-TOF
can also be used to check for sequence errors that may occur while inputting sequences. Such a QC method is an absolute
requirement for sequence dependent experiments, such as PCR, cloning and sequencing. It can also be used to check
whether an oligo has been modified correctly. CE or HPLC analysis cannot be used to check modifications. MALDI-TOF is
also used to check for the presence of truncated oligonucleotides and salt contamination.
A MALDI-TOF mass system is most suitable for the QC of oligonucleotides less than 50 bases long. Longer oligonucleotides
(> 50 mer) cannot be ionized effectively (100%) by the laser, therefore they cannot be easily detected and therefore
will show a poor detection signal that may fail QC. At Bioneer any oligo greater than 50 bases in length are checked
for quality by PAGE. PAGE QC data sheets are provided with each oligo > 50 bases.
The Bioneer QC system
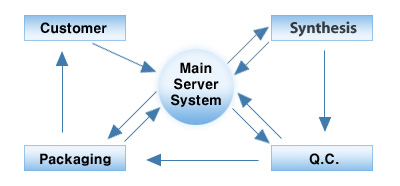
The customer order data is initially saved on the main server system and then transferred to synthesis. Following
synthesis, oligonucleotides are spotted on MALDI-TOF mass plates using a proprietary, fully automated, 384-well sample
OD quantification/ dispensing robot developed by Bioneer. The MALDI plates are then transferred to QC Division.
Any transfer of oligo samples and plates between different divisions and/or equipment requires the bar-code on each
sample racks to be checked by a production specialist to confirm the oligo data. Bar coding ensures compliance and
allows related divisions to easily retrieve important oligo data from the main server system.
After receiving the oligonucleotides and all the related information, the QC department checks the quality of the
oligonucleotides. The mass spectrum of each oligo is saved and the QC program checks whether the oligonucleotides have
been synthesized appropriately. Upon completion the final QC data is transferred to the main server.
The Bioneer QC Program is also used to confirm oligo contaminants (including truncated oligonucleotides) present in
the MALDI spectra. Another key advantage of Bioneer's QC system is its ability to automatically insert mass spectra
(0.6X mass ~ 1.3X mass) of each oligo into the oligo data sheet. Mass spectrum for all ordered oligonucleotides will be
provided to customers at no extra charge.
Please note that since the Sequenom Spectrocheck system, which is applicable for some SNP users employs its own program
for oligo QC, $100 USD will be charged for every 96 samples if this QC system is required. Mass spectrum data of examined
samples can be provided on a CD if required.
MALDI data is delivered from QC to the main server, and subsequently all the related information is delivered to
Packaging so that the correctly synthesized samples will be delivered in the appropriate format as requested by the
customer. Bioneer delivers oligonucleotides in a selection of different tubes, 96-well plates, or 384-well plates as
per the customers' preferences. After packaged completely, all the oligonucleotides will be shipped by FedEx or UPS,
and via their tracking systems, customers may monitor the exact place where the ordered oligonucleotides are in
transportation.
In QC, data on all failed samples is automatically returned to synthesis and the re-synthesis of the failed
oligonucleotides proceeds whilst QC examines the failed oligo further. This rapid exchange of related information is
a key to Bioneer's rapid oligo turnaround time.
HPLC
HPLC Analysis of Oligo Purity
Reverse Phase HPLC
At Bioneer Reverse Phase HPLC is mostly used to QC of intermediates or single stranded DNA produced in the oligo
synthesis process. It is a simple QC technique for modified oligo with hydrophobic groups. Reverse Phase is faster
and cheaper than Ion Exchange methods and requires less sample.
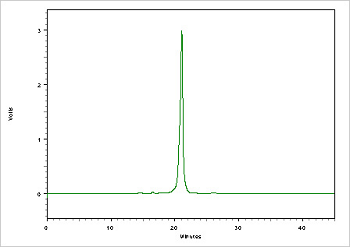
Figure 4. Example of oligo (26 mer) purity analysis using a Reverse Phase (C-18) Column.
Purity Analysis using Ion-exchange Chromatography Method (using Anion-exchange column)
HPLC, equipped with a DIONEX's DNAPac column, is used to QC of oligonucleotides, in particular - Decoy oligonucleotides.
The high resolution capability of Ion-exchange can easily separate single stranded DNA and double stranded DNA.
At Bioneer Ion-exchange chromatography is commonly used to QC decoy oligonucleotides, and plays a key role in QC
confirmation with strict QC standards required for gene therapy.
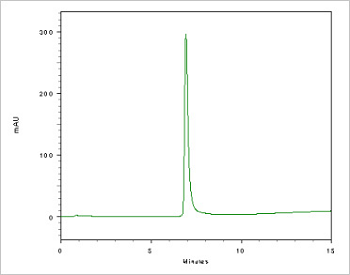
Figure 5. Double stranded DNA (DS DNA) test. Double stranded oligonucleotides are used for decoy or EMSA
experiments. Prior to any experiments, the formation of DS DNA must be checked. For decoy experiments used in the
development of new drugs, it is necessary to check the ratio of DS DNA in the decoy. In order to guarantee the medical
efficacy, the medicine should be formed in decoy like API from the start of drug development. DS DNA confirmation is
an FDA requirement.
GC
Product Purity Test Under Gas Chromatograph
Gas Chromatograph (GC) is used to QC for solvent content in Decoy oligonucleotides and S-oligos used in gene therapy.
Prior to administering any oligo based drug to humans it is vitally important to check for the presence of residual
organic solvents that may remain after synthesis and purification. Solvent content may compromise efficacy and cause
unwanted side effects. The types of residual organic solvents that may be present include acetonitrile, pyridine and
toluene etc. Concentrations should be minimally less 0.1%.
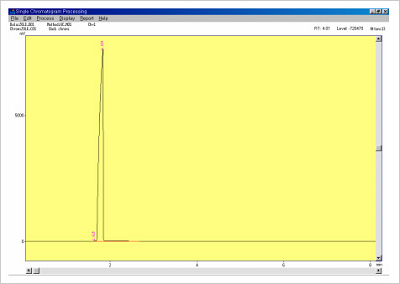
Figure 6. Standard solvent data.
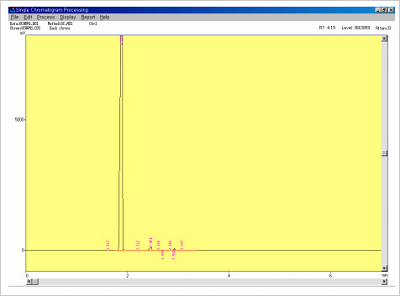
Figure 7.Solvents confirmation data in antisense oligonucleotide
NMR – Spectroscopy
Nuclear Magnetic Resonance (NMR) spectrometer plays a very important role in understanding 3-dimensional structures
of molecules. With increasing interests in the structure of biological materials, the use of NMR spectrometer is
expanding into new areas, such as drug development, DNA analysis, human genomic and proteomic research and so on.
NMR is commonly used to determine physical structure at the molecular level.
At Bioneer NMR is used for 31P-NMR measurement to compare typical frequency values for phosphates present in DNA
backbones. By comparing actual frequencies with theoretical it is possible to check the state and purity of phosphates
in synthetic oligonucleotides.
Heavy Metal Testing
For antisense and decoy oligonucleotides that are directly injected into animals or humans as medicines in the
pre-clinical or clinical phase, it is necessary for check for heavy metal groups that may influence the efficacy
or may cause side effects. The types of heavy metals that require QC may differ in each oligo. Inductively Coupled
Plasma-Optical Emission Spectrometers (I.C.P), Atomic Absorbance Spectrophotometers (AAS) and I.C.P Mass Spectrometers
are routinely employed to QC oligonucleotides for heavy metal groups. Upon request, Bioneer's oligonucleotides can be
checked quantitatively/qualitatively for metals such as Lead, Nickel and Fe etc.
Water Content Analysis
Bioneer can also QC oligonucleotides for water content. A Sartorius’ Water Content Measurement instrument (MA-30) is
employed to measure water contents that may remain in synthesized antisense oligo following the final drying step of
the oligo purification process.
Bioburden Testing
Bioneer confirms the sterility of an aseptic oligo production environment by routinely conducting microbial testing
of the water used in the synthesis process and final aliquoting steps. Susceptible areas of potential microbial
contamination in the synthesis process and the environment, including operators are also checked periodically.
Prevention ensures that the final oligonucleotides will be proven to be safe and free from microbial contaminations.
Endotoxin test
Bioneer also utilizes a Kinetic Chromogenic Analysis (KCA) method to confirm that the oligonucleotides are free of
any exothermic materials. Generally exothermic materials present in injectable therapeutics are endotoxins from
microbial contamination, especially from Gram negative bacterial contamination and must be avoided.
Kinetic Chromogenic Analysis (KCA) is based on an enzyme linked color reaction (limulus Amoebocyte Lysate reaction).
The presence of endotoxins is quantified by measuring color of the reaction against known standards. Many samples
can be quantified simultaneously using a standard micro-plate reader. KCA is a fast, cost-effective and short
measurement. With such a method, Bioneer only provides oligonucleotides with less than 0.25 EU/ml for therapeutic
applications.
OVERVIEW of BIONEER QA/QC
All oligonucleotides synthesized by Bioneer are purified using the Bio-RP purification system. This uses the oligo
purification cartridge (OPC) technology and generates oligonucleotides that are 85% pure. This service is available
free of cost on all oligonucleotides. We also offer HPLC and PAGE purification.
Bio-RP Purification
The Bio-RP cartridge functions on a basic trityl on selection. In addition to a de-salting, it removes most of the
truncated products. This technology yields oligonucleotides that are superior in purity than that obtained by standard
ethanol precipitation or gel filtration.
PAGE Purification
PAGE purification is recommended for oligonucleotides between 50-100 bases. This yields oligonucleotides with purity
levels over 95% purity. Subsequently yield obtained is lower than other purification techniques.
HPLC Purification
HPLC is recommended for both modified and unmodified oligonucleotides up to 50 bases in length. Our HPLC purifications
always start with a trityl on purification, and are often followed by a second full gradient trityl off HPLC. This is
our Double RP-HPLC Purification process that gives an extra level of purification. In some cases, the initial trityl-on
purification is entirely sufficient and a cartridge is utilized in the subsequent trityl-off oligo processing.
This RP-HPLC purification process can yield highly pure material, especially when the oligonucleotide is not complex.
When synthesizing compounds that require post-synthetic modification, such as dye labeling, we will perform an
additional RP-HPLC purification.
GUARANTEED MINIMUM YIELD
Yield will vary depending on base composition, purification method, oligo length, and synthesis scale.
To better serve our customers, Bioneer has set standards for guaranteed yields. In addition, our Quality Control
(QC) department checks each and every oligo that Bioneer ships out by MALDI-TOF or Sequenom MassARRAY Oligo Check.
QC data is included with every shipment.
* Please note OD values apply to tube oligonucleotides, and nmole values apply to plate oligonucleotides.
* Please note the yield of oligonucleotides with modifications and PAGE or HPLC purification will be decreased by 15 to
20%.
| BIO-RP(Cartridge) |
20mer |
nmol |
9 |
19 |
37 |
134 |
1333 |
2005 |
| OD |
2 |
4 |
8 |
29 |
288 |
433 |
| 30mer |
nmol |
6 |
12 |
25 |
90 |
892 |
1341 |
| OD |
2 |
4 |
8 |
29 |
288 |
433 |
| 40mer |
nmol |
4 |
9 |
19 |
67 |
667 |
1002 |
| OD |
1.5 |
4 |
8 |
29 |
288 |
433 |
| 50mer |
nmol |
3 |
7 |
15 |
54 |
534 |
803 |
| OD |
1.5 |
4 |
8 |
29 |
288 |
433 |
| 60mer |
nmol |
2 |
6 |
12 |
45 |
444 |
668 |
| OD |
1 |
4 |
8 |
29 |
288 |
433 |
| 70mer |
nmol |
|
5 |
11 |
38 |
382 |
574 |
| OD |
|
4 |
8 |
29 |
288 |
433 |
| BioRP + Modification |
20mer |
nmol |
7 |
15 |
30 |
107 |
1066 |
1604 |
| OD |
2 |
3 |
6 |
23 |
230 |
346 |
| 30mer |
nmol |
5 |
10 |
20 |
72 |
714 |
1073 |
| OD |
2 |
3 |
6 |
23 |
230 |
346 |
| 40mer |
nmol |
3 |
7 |
15 |
54 |
534 |
802 |
| OD |
1.5 |
3 |
6 |
23 |
230 |
346 |
| 50mer |
nmol |
2 |
6 |
12 |
43 |
427 |
642 |
| OD |
1.5 |
3 |
6 |
23 |
230 |
346 |
| 60mer |
nmol |
1 |
5 |
10 |
36 |
355 |
534 |
| OD |
1 |
3 |
6 |
23 |
230 |
346 |
| 70mer |
nmol |
|
4 |
9 |
30 |
306 |
459 |
| OD |
|
3 |
6 |
23 |
230 |
346 |
| PAGE |
20mer |
nmol |
|
3.0 |
4 |
19 |
153 |
231 |
| OD |
|
0.7 |
0.9 |
4 |
33 |
50 |
| 30mer |
nmol |
|
2.0 |
3 |
12 |
102 |
155 |
| OD |
|
0.7 |
0.9 |
4 |
33 |
50 |
| 40mer |
nmol |
|
1.0 |
2 |
9 |
76 |
116 |
| OD |
|
0.7 |
0.9 |
4 |
33 |
50 |
| 50mer |
nmol |
|
0.7 |
1 |
6 |
50 |
74 |
| OD |
|
0.5 |
0.8 |
3 |
27 |
40 |
| 60mer |
nmol |
|
0.7 |
1 |
3 |
34 |
49 |
| OD |
|
0.5 |
0.8 |
2 |
22 |
32 |
| 70mer |
nmol |
|
0.7 |
1 |
3 |
23 |
33 |
| OD |
|
0.5 |
0.8 |
2 |
17 |
25 |
| HPLC |
20mer |
nmol |
|
3.0 |
4 |
28 |
185 |
277 |
| OD |
|
0.8 |
1 |
6 |
40 |
60 |
| 30mer |
nmol |
|
2.0 |
3 |
18 |
123 |
186 |
| OD |
|
0.8 |
1 |
6 |
40 |
60 |
| 40mer |
nmol |
|
1.0 |
2 |
13 |
92 |
140 |
| OD |
|
0.8 |
1 |
6 |
40 |
60 |
| 50mer |
nmol |
|
0.7 |
1 |
6 |
65 |
92 |
| OD |
|
0.8 |
1 |
3 |
35 |
50 |
| 60mer |
nmol |
|
0.7 |
1 |
4 |
46 |
61 |
| OD |
|
0.8 |
1 |
3 |
30 |
40 |
| 70mer |
nmol |
|
0.7 |
1 |
3 |
33 |
40 |
| OD |
|
0.8 |
1 |
3 |
25 |
30 |
Q1. How should I store my oligo?
Normally, oligos should be stable at -20°C and can be stored at that temperature for more than a year. Although stable
in solution, oligos will be degrade if the storage solution is contaminated with nucleases. Therefore, we recommend
that oligos be stored in the dried form. If you want to store oligos in solution, it is best to aliquot the oligo
into several tubes and store them separately. Oligos can also be subject to degradation due to the 'Freezing and
Thawing Effect' when the oligo solutions are frozen and thawed repeatedly. For storage of oligodeoxyribonucleotide
(DNA), pH value should be maintained at neutrality. Under acidic conditions, DNA can become depurinated. On the
other hand, the phosphodiester bond of oligoribonucleotides (RNA) can be hydrolyzed under basic conditions.
Q2. How should I resuspend my oligo?
For long-term storage we recommend that the oligos be dissolved in a buffer, such as TE (10 mM Tri-HCl, 1 mM EDTA,
pH 8.0), instead of just sterilized water. Once resuspended, oligos should be kept frozen at -20°C. Since some
oligos may not be easily dissolve in sterilized water the addition of NaOH does help dissolve oligos in water.
Q3. If oligos were left at room temperature for more than a week, would they still work?
Once dried, oligos are supposed to have tremendous stability. Even in solution, they are reasonably stable.
Therefore, in most cases, without contamination by materials which can cause decomposition of oligos,
they should still work well, even if they were left at room temperature for more than a week.
Q4. Do I have to treat fluorescent dye modified oligos differently in storage and handling?
If exposed to light, fluorescent dye modified oligos are more fragile than unmodified oligos and their
fluorescence intensity will decrease over time. To maintain their fluorescence efficiency, fluorescent dye
modified oligos should be stored in the dark at -20°C.
Quantity and Concentration
Q5. How does Bioneer quantify my oligo?
The quantity of oligo we provide is based on its UV optical density (OD) measured at a wavelength of 260 nm.
Q6. How do I calculate the oligo quantity from the measured OD value?
The quantity of oligos is often described in O.D. units which actually express light absorbance. 1 O.D.
corresponds to the amount of oligo in a 1 mL volume that results in an optical density of 1 in a 1 cm path-length
cuvette. This corresponds to approximately 33 µg of oligo, although it varies for each particular oligo depending
on its sequence. The concentration of an oligo of known sequence can be calculated since it is known that the
extinction coefficient (in a 1 cm path-length cuvette) for each of the bases at 260 nm is
dG : 11.7 mL/µmole
dC : 7.3 mL/µmole
dA : 15.4 mL/µmole
dT : 8.8 mL/µmole
For any given oligo, multiply the number of times each base appears by its extinction coefficient. Then add the
resulting four numbers to obtain the extinction coefficient (e) for the entire oligo. The concentration (C) can
then be calculated from the equation (for a 1 cm path-length cuvette):
O.D. = e * C
Q7. If the O.D. value of an 18 mer oligo containing 3dG, 4dC, 5dA and 6T is 0.7, how much oligo is there?
First, calculate the extinction coefficient (e) for the entire 18-based oligo as follows:
e = 11.7 x 3 + 7.3 x 4 + 15.4 x 5 + 8.8 x 6 = 194.1 (mL/μmole)
Next, obtain the concentration of oligo in the cuvette from the equation O.D. = e * C
C = 0.7 /194.1 = 0.0036 (mmole/mL) = 3.6 (nmole/μL)
Finally, the amount of 18-based oligo having 0.7 O.D. is 3.6 nmole.
Q8. How do we calculate the molecular weight of an oligonucleotide?
The molecular weight of an oligo can be calculated with the following equation:
M.W. = NA * 249.2 + NC * 225.2 + NG * 265.2 + NT * 240.2 + (oligo length-1) * 63.98 + 2.02
NA = Total # of A ; NC = Total # of C ; NG = Total # of G ; NT = Total # of T
Q9. How do I convert oligo quantity expressed in nmole into weight?
Normally, the amount of synthetic oligonucleotide is described in number of moles, usually nmole.
The amount of oligo can easily be calculated from the following equation:
Amount of oligo (ng) = Molecular Weight (M.W. in g) X Number of moles (nmole)
Q10. When I do not know the exact base composition, is there any method to quantify the synthesized oligo?
Approximately - a single stranded oligo with 1 O.D. value contains 33 µg while double stranded oligo contains 50μg.
For short oligos, however, there would be big deviations from the above values.
Q11. How do I measure Tm of the synthesized oligo?
Tm (melting temperature) refers to the temperature where 50% of oligonucleotides exist in duplex form and the rest
in single-strand form.
There are several ways to calculate Tm. At Bioneer, we use the nearest-neighbor method (PNAS 83, 3746-50).
It is believed that the effect of hybridization is different for every sequence and that through thermodynamic
measurements; you can estimate Tm values more accurately. For example, the sequences of 5'-GC-3' and 5'-CG-3'
are different in thermodynamic measurements.
The method for nearest-neighbor calculation is as follows:
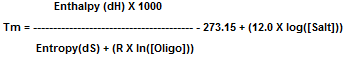
Through thermodynamic measurements, enthalpy and entropy values are determined between 2 bases. [Salt] is the
concentration of monovalent cations and [Oligo] is the oligo concentration. R is the gas constant (1.987 cal•K-1mole-1).
Bioneer's Tm value is calculated by the nearest-neighbor method with 50mM for salt concentration and 1 nM for oligo
concentration.
Please note that there are other ways of estimating the Tm. For oligos shorter than 15 mer, the Wallace rule can
be used:
Tm = 2°C (A + T) + 4°C (G + C)
Another estimation method based on the GC content for long sequences is:
Tm = 81.5 + 0.41(%GC) - 500/L + 16.6 log[M]
(L; oligonucleotide length, [M]; monovalent cation concentration)
However, these methods do not consider the base stacking effect and usually the estimation is not as accurate as the
nearest neighbor method. Nonetheless, there are still some disadvantages in the nearest neighbor method for 60-70 or
under 15 bp estimation.
Bioneer provides the Tm values of every oligo, but the values are estimations and we cannot guarantee the exact values.
Therefore, the Tm value should be used only as a reference.
If the experiment does not yield anticipated results, it is recommended to lower the annealing temperature by 4-5 degrees
from the Tm value. If there are many non-specific products, trial-and-error approach should be taken to obtain the
optimum annealing temperature.
Q12. Why are there differences in Tm value that Bioneer provided and mine?
The TM Calculator that BIONEER uses is different from, and more accurate than, the more commonly used calculators
based on the Wallace rule.
Q13. What is the method for adjusting the oligonucleotide concentration?
On the data sheet that Bioneer provides for each oligo, the volume of TE buffer or distilled water necessary to
make a 100 pmole/μL oligo solution appears by the "volume for 100 pmole/μL" heading.
For example, if 189.0 is indicated on the sheet, add 189 μL of TE buffer to the tube.
Such a solution would be 100 μM in concentration:
100 pmole/μL = 100 x 10-12 mole/10-6 L
= 100 x 10-6 mole/L
= 100 x 10-6 M
= 100 μM
Q14. Unit conversions
System of scientific units:
10-1 = deci [d]
101 = deca [da]
10-2 = centi [c]
102 = hecto [h]
10-3 = milli [m]
103 = kilo [k]
10-6 = micro [μ]
106 = mega [M]
10-9 = nano [n]
109 = giga [G]
10-12 = pico [p]
1012 = tera [T]
10-15 = femto [f]
1015 = peta [P]
10-18 = atto [a]
1018 = exa [E]
10-21 = zepto [z]
1021 = zetta [Z]
10-24 = yocto [y]
1024 = yotta [Y]
----------------------------------------------
Example for unit exchange of oligonucleotides
1 pmole/μL
= 1x10-12 mole / 1x10-6 L
= 1x10-6 mole / L
= 1 μmole / L
= 1 μM
Synthesis and Order
Q15. How are oligonucleotides synthesized?
The most popular method for synthesizing oligonucleotides is to form natural 3’-5’ phosphodiester bonds between monomers
by using ‘phosphite triester’ protocols. ß-cyanoethyl phosphoramidites, the building monomers, were developed by Koster
and used most often to synthesize oligonucleotides (Nucl. Acids Res. 1984, 12, 4539 ; Tetrahedron Lett. 1983, 24,5843).
Through the ‘phosphite triester’ method using ß-cyanoethyl phosphoramidite, high coupling efficiency is achieved ( > 98%)
and the time consumed for coupling is much shorter than that of other methods of oligo synthesis. Moreover, since the
monomers, ß-cyanoethyl phosphoramidites, are quite stable prior to the activation, which is necessary for oligo
synthesis, and means they can be stored for a long period of time.
The oligonucleotide is synthesized while attached covalently to a solid support. Excess soluble protected nucleoside
β-cyanoethyl phosphoramidites and coupling reagent can drive the reaction near to completion. Among the solid supports,
controlled pore glass (CPG), which consists of a glass matrix prepared uniformly with pores of defined size, has been
used predominantly over the last few years.
The whole synthesis of oligonucleotides can be accomplished by the chain reactions where four different reaction cycles
- deblocking, coupling, oxidation and capping are performed repeatedly (Fig. 1).
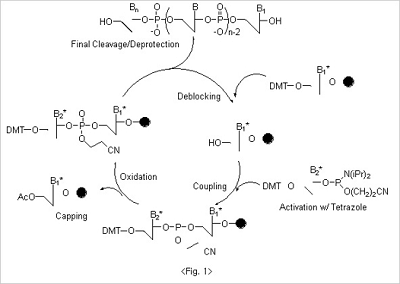
Deblocking
In the first step in of synthesis - deblocking - the 5’ protecting group, DMT, is cleaved from the CPG. For deblocking,
acidic condition is necessary, and trichloroacetic acid (3% in dichloromethane) is used in most of cases. It is reported
that oligos can be depurinated in acidic conditions, especially more severe for adenosine. Since trichloroacetic acid
is very acidic (pKa : ~1.5), deblocking solution with trichloroacetic acid should not be left too long in the reaction.
Instead of trichloroacetic acid, dichloroacetic acid, which is less acidic than trichloroacetic acid, can be used for
deblocking to avoid the depurination problem in certain cases. Since the DMT cation, which is produced after
deblocking cycle, shows a very strong orange color, it can be used to monitor the coupling efficiency by measuring
its light absorbance.
Coupling
The 5'-hydroxyl group on the CPG, which is exposed after the deblocking step, is coupled to the nucleoside β-cyanoethyl
phosphoramidites to form triphosphite ester which is subsequently oxidized to a phosphotriester bond. For nucleoside
β-cyanoethyl phosphoramidites, to avoid the unwanted side reaction during the whole oligo synthesis, exocyclic amino
groups in base moiety are protected to result amide structure. Benzoyl groups are used for both adenosine and
cytidine. On the other hand, isobutyryl group is used for guanosine base protection. Since thymidine doesn't have
exocyclic amine group in base there is no need for extra protection. 5'-Hydroxyl groups are protected with DMT
for all nucleoside βcyanoethyl phosphoramidites (Fig. 2).
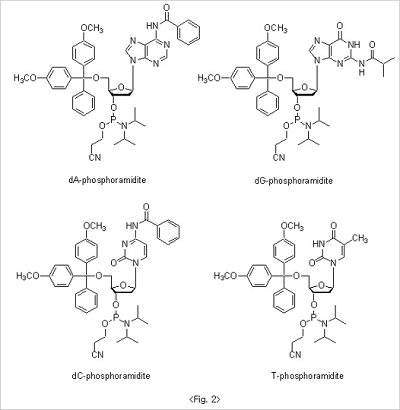
Since nucleoside β-cyanoethyl phosphoramidites are quite stable under normal conditions, they cannot react directly
with a free 5’ hydroxyl function on a growing chain. They must first be activated by treatment with an activator
usually a type of weak acid. Among a variety of candidates, tetrazole has shown a great efficiency and has been
used as a standard activator. Tetrazole has been thought to play a dual role: it protonates the diisopropylamino
group of the phosphoramidite function; and then comes in as a nucleophile, generating a very reactive
tetrazolophosphane intermediate. Coupling reactions with these activated nucleoside phosphoramidite reagents are
very fast (less than 2 minutes) and are almost quantitative.
Oxidation
The newly formed phosphite internucleotide linkage is unstable and susceptible to both acidic and basic cleavage.
Therefore, the trivalent phosphite triester is oxidized to a stable pentavalent phosphate triester. Iodine is used as
a mild oxidant in basic tetrahydrofuran solution with water as the oxygen donor. The reaction is extremely fast,
being quantitative in 30 seconds.
Capping
Since the coupling reaction cannot be quantitative in a finite time period, a small percentage of truncated sequences
are produced at every coupling step. If these failure sequences were allowed to react further, it would be difficult
to isolate the product from the sequence mixture. This problem is overcome largely by capping the remaining free
hydroxyls through acetylation.
Acetylation is achieved with the strong acetylation reagent which forms on reaction of equimolar amounts of acetic
anhydride and N-methylimidazole. The reaction is almost quantitative in 30 seconds.
After oxidation, the nucleotide addition cycle is complete. Oligonucleotide synthesis can continue removing the DMT
group at the 5’ -end of the growing chain and repeating another cycle of nucleotide addition.
At the end of whole synthesis of oligonucleotides, cleavage from support and simultaneous base and phosphate
deprotection are achieved by treatment with concentrated ammonium hydroxide.
Q16. Standard oligo structure.
Q17. What are base limitations on each synthesis scale?
0.025 μmole synthesis scale: 15 – 60 mer
0.05 μmole synthesis scale: 10 – 75 mer
0.2 μmole synthesis scale: 5 – 110 mer
1.0 μmole synthesis scale: 5 – 130 mer
10 μmole synthesis scale: 5 – 50 mer
15 μmole synthesis scale: 5 – 50 mer
Q18. When I ordered the 50 nmole scale, I got less than 50nmoles. What happened?
50 nmole scale synthesis of oligos doesn’t mean we can guarantee 50 nmole of final oligos. Instead, 50 nmole scale
refers to the loading amount of solid support used at the beginning of oligonucleotide synthesis. Since oligos are
usually ordered by the reaction scale not the final yield, the amounts of oligos which customers could get is naturally
less than ordered. The final yields can vary with oligo length, base composition and coupling efficiency.
Q19. Can you make the oligos having a high percentage of "G" residues?
It is known that oligos having a high percentage of “G” residues are difficult to synthesize, especially if sequence
contains several “G” in a row. It is also reported if there are “G”s existed four or more in a row, oligos tend
to aggregate and form “guanine tetraplex”. (Poon and MacGregor, Biopolymers, 1998, 45, 427-434) By substitution of
inosine for some of “G”, the formation of “guanine tetraplex” can be disrupted.
Q20. Do you provide oligoribonucleotide (RNA) synthesis?
Yes, we do. We can offer oligoribonucleotide with 2’ -OH and/or 2’ -O-methyl structure at the desired site.
We can also synthesize the chimeric oligos which have DNA and RNA structures mixed.
Q21. Does the oligo synthesized have phosphate group at 5’ or 3’ position?
If not ordered separately, the oligos synthesized do not contain phosphate group at 5’ or 3’ position.
If you want to have oligo phosphorylated at 5’ or 3’, you should specify 5’ or 3’ phosphorylation modification
when ordering.
Q22. What are the symbols denoting degenerate bases?
R: a or g
Y: c or t
M: a or c
K: g or t
S: g or c
W: a or t
V: a, c, or g
H: a, t, or c
B: g, t, or c
D: g, a, or t
N: a, c, g, or t
Purification
Q23. How do you purify the oligos synthesized?
To purify the synthesized oligonucleotides, Bioneer adopts several methods such as desalting, OPC, HPLC and PAGE.
Desalting
After completion of oligo synthesis, to purify oligos as a natural DNA structure, protection groups must be deprotected
first. By treatment of concentrated ammonium hydroxide the synthesized oligos are cleaved from the solid support,
b-cyanoethoxy, the protecting group for the phosphodiester bridge, can be removed and finally protecting groups
(benzoyl and isobutyl) for base moiety can be removed to result in natural DNA structure. After all necessary
deprotection processes are complete the oligo mixtures contain several small organic materials which must be removed.
The process that all the unnecessary organic materials are removed is commonly referred to as “desalting”.
The desalting purification can be done by using the Sep Pak columns that contain reverse-phase silica gel.
Although desalting purification can successfully remove all the unnecessary organic impurities, it is not capable
of removing the truncated oligo impurities efficiently that are generated inevitably during synthesis even
in very tiny amounts (less than 2% for each coupling step). The desalted oligos, however, can be used without
serious problems in most biological applications like PCR detection.
OPC
If the oligo synthesis is complete on “trityl-on” mode, the final N-mer oligo contains a 5’ -DMT group and the truncated
oligos do not. Since DMT group has a strong lipophilic character, the oligo with 5’ -DMT group has an affinity to RP
resin which is usually used for oligo purification. By using the fact that a strong affinity between an RP resin and
oligo with 5’ -DMT group exists but the truncated oligos do not contain DMT group, we can successfully separate the
N-mer oligo from the unwanted truncated oligo impurities.
HPLC
For the applications such as cloning, site directed mutagenesis or quantitative gene detection, oligos of higher purity
are preferred to get the satisfactory results. Since desalted or OPC purified oligos might not be sufficient in such
cases, HPLC purification has been commonly used for that purpose. Either anion-exchange or reverse-phase can be used
for oligo purification. HPLC with an anion-exchange column delivers 95~98% purification efficiency and is adequate
for purification of oligos up to 35 mer. HPLC with a RP column shows similar purification efficiency to
anion-exchange, but since the purification efficiency of HPLC depends strongly on the oligo length, the long-length
oligos (> 35 mer) might not be purified efficiently with HPLC purification.
PAGE
For the purification of long-length oligos (up to 100 mer), Bioneer recommends PAGE purification which uses cross-linked
polyacrylamide gel as purification matrix. Though PAGE shows high purification efficiency (> 98%), it has a couple of
shortcomings in that extra processes such as extraction and desalting are required following PAGE and subsequently
results in a decrease in purification yields.
Q24. How is long-mer oligo for microarray purified?
We recommend PAGE purification for long-mer purification. Normally, long oligos (> 50 mer) are recommended to
be purified with PAGE.
Q25. Oligo modifications.
5’ Amine and 3’ amine: The presence of a primary aliphatic amine group at the terminus of an oligonucleotide allows
the post -synthesis attachment of a number of amine reactive molecules. The amino modified oligonucleotides can be
used for the immobilization of the oligo on a matrix (oligonucleotide-based microarrays).
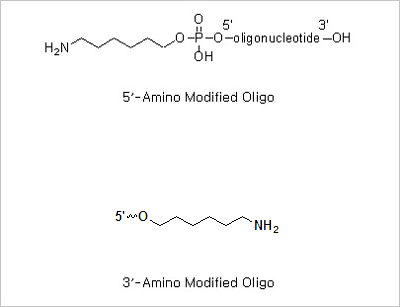
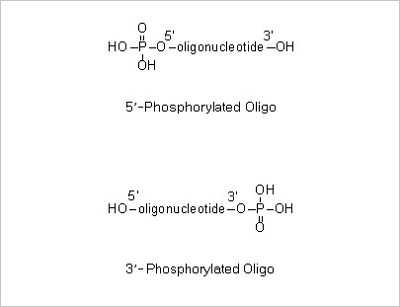
5’ Phosphorylation and 3’ Phosphorylation Modifications: 5’ -Phosphorylated oligonucleotides are commonly
used in site directed mutagenesis and linker insertion.
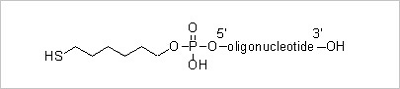
5’ Thiol Modification: Thiol modification at the 5’ end of an oligonucleotide allows further derivatization of
thiol reactive molecules. The thiol moiety in the oligonucleotides can be conjugated to a variety of different
fluorophores for DNA sequencing and hybridization.
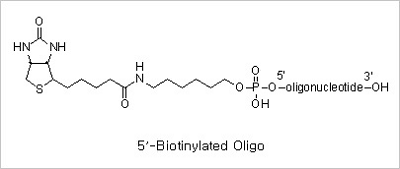
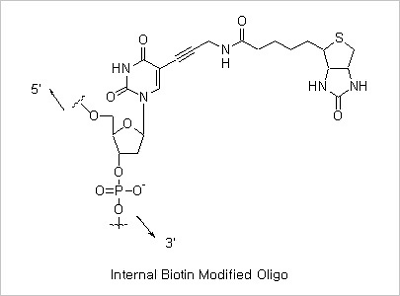
5’ Biotin and Internal Biotin-dT Modifications: Biotinylated oligonucleotides are used for a number of applications,
which include colorimetric detection of DNA and solid phase capture by Streptavidin coated magnetic beads for use
in restriction mapping, genomic walking and differential display.
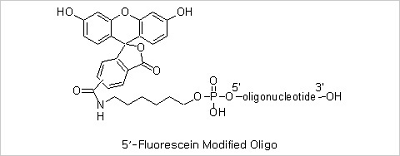
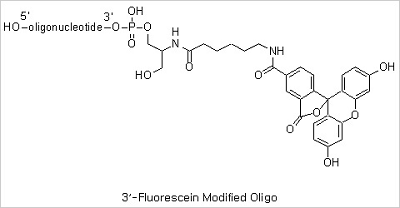
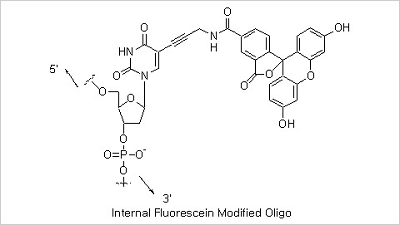
5’ Fluorescein, 3’ Fluorescein and Internal Fluorescein Modifications: Fluorescent dye-labeled oligonucleotides
are widely used in automated DNA sequencing, quantitative PCR, and in situ hybridization reactions. Among the variety
of fluorescent dyes, fluorescein (excitation/emission maxima ~494/520 nm) is one of the most commonly used fluorophores
in labeling and detection of biomolecules. In addition to its relatively high absorptivity and excellent fluorescence
quantum yield, fluorescein has an excitation maximum that closely matches the 488 nm spectral line of the argon-ion
laser, making it the predominant fluorophore of confocal laser scanning microscopy and flow cytometry applications.
Fluorescein, however, has several drawbacks, including a relatively high rate of photobleaching, as well as
pH-sensitive fluorescence (pKa ~6.4) that is significantly reduced below pH 7. In spite of these drawbacks
fluorescein has still been the mostly used fluorescent dye due to its good availability.
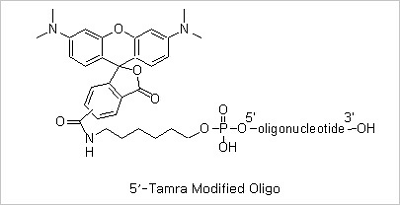
5’ TAMRA and 3’ TAMRA Modifications: Rhodamines are considered more photostable fluorescent dyes compared to fluorescein.
The spectra of Rhodamines’ are not affected by changes in pH between 4 and 10, an important advantage over the
fluoresceins for many biological applications. The most common member of these rhodamine groups is tetramethylrhodamine
(TAMRA) which is an important fluorophore for oligonucleotide labeling and automated DNA sequencing applications.
The fluorescence quantum yield of TAMRA conjugates are usually only about one-fourth that of fluorescein conjugates.
However, because TAMRA is readily excited by the intense 546 nm spectral line form mercury-arc lamps used in most
fluorescence microscopes and is intrinsically more photostable than fluorescein, TAMRA conjugates often appear
to be brighter than the corresponding fluorescein conjugates. TAMRA is also efficiently excited by the 543 nm
spectral line of the green He-Ne laser, which is increasingly being used for analytical instrumentation. TAMRA
conjugates are not well excited by the 568 nm line of the Ar-Kr mixed gas laser used in many confocal laser
scanning microscopes.
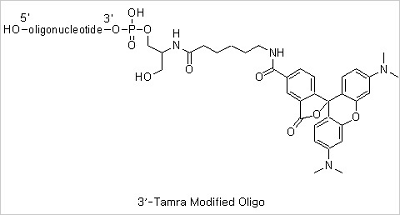
5’ Cy3 and 5’ Cy5 Modifications: The maximum wavelengths of absorption/emission for Cy3 and Cy5 are 550/570 and 649/670
nm respectively.
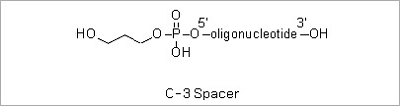
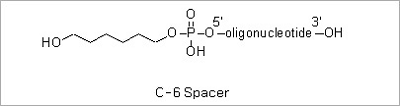
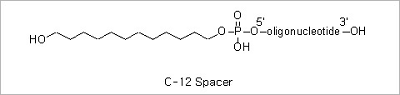
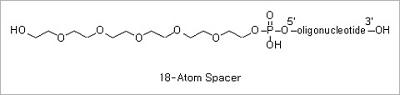
Spacer Modifications: Incorporation of a phosphoramidite containing a variety of spacers between the oligonucleotide
and a subsequently attached label may be necessary in situations where a label interferes with oligonucleotide
hybridization.
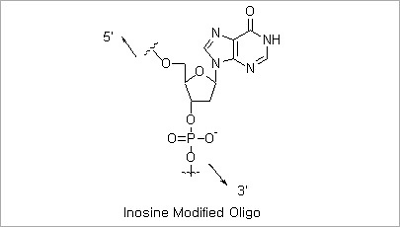
Inosine Modification: Deoxyinosine forms stable base pairing with an order of stability of I:C > I:A > I:T = I:G.
Oligonucleotides containing inosine may be used in applications where the detection or analysis of distinct but similar
DNA sequences by probe hybridization or PCR is carried out.
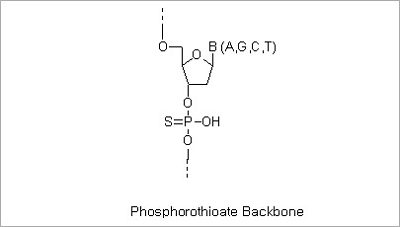
Phosphorothioate Modification: The modified “backbone” of an S-Oligo is resistant to degradation by most endo- and
exonucleases. This property allows increased intracellular effectiveness of antisense oligonucleotides. The replacement
of the inter-nucleotide phosphate groups with phosphorothioate groups (substituting one of the oxygen atoms of
phosphate group with a sulfur atom) can be made in the entire of partial sequences of oligo depending on the
customers’ needs.
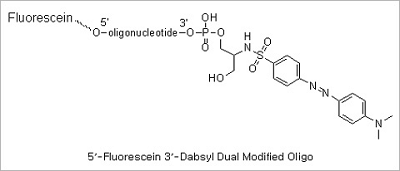
Dual modification (5’ Fluorescein 3’ DABCYL & 5’ Fluorescein 3’ TAMRA modifications): Dual probe oligonucleotides are
used in Real-time PCR. They are either cleaved in the reaction (as TaqMan probes) or undergo a conformation change
in the presence of a complementary DNA target (as Molecular Beacons). Many probe designs employ oligonucleotides that
form reversible stem-loop configurations. In each case, the probes signal the reaction occurrence by eliminating
the quenching influence on a donor fluorophore.
5’ nuclease assay for PCR monitoring (TaqMan probes):
The 5’ nuclease assay was developed to allow the real-time monitoring of the PCR reaction. The assay utilizes the
5’ ->3’ exonuclease activity of Taq DNA polymerase to monitor the ongoing reaction. Taq DNA polymerase can cleave 5’
terminal nucleotides from the strand of DNA that it displaces while synthesizing a new strand. The cleavage primarily
occurs at the junction between the displaced single stranded portion and the double stranded paired part of the DNA
strand. This results in the release of mono- and oligonucleotides from the 5’ end of the displaced DNA strand.
That property of Taq DNA polymerase was used to monitor the reaction. The FRET DNA dual probes in the 5’ nuclease
assay are short oligonucleotides, complementary to the target sequence in the amplified DNA and 3’ -modified
so that they cannot be extended on the 3’ end. They are labeled with fluorophores at both the 3’ and 5’ ends.
The reporter dye is positioned at the 5’ end, and the quencher is positioned on the 3’ end of the probe. With
the probe intact, the reporter dye fluorescence is quenched. In the reaction, the probe hybridizes to the
target sequence. During PCR, it is cleaved by the 5’ nuclease activity of Taq DNA polymerase. The cleavage
separates the quencher from the reporter and restores its fluorescence. Two probes were used, one for the
normal sequence and the other complementary to the sequence containing a 3-bp deletion. Each probe had a
different reporter fluorophore at the 5’ end and a common quencher dye attached to the seventh nucleotide
from the 5’ end. The identity of the target DNA was determined from the fluorescence emission spectrum.
Molecular Beacons:
Strictly speaking, the fluorescence quenching in molecular beacons that use DABCYL is not FRET-related because it does
not satisfy the spectral overlap criteria. However, the FRET-based quenching can also be used. Molecular beacons found
applications in PCR soon after their introduction as hybridization probes. They can monitor an ongoing reaction and
discriminate alleles in real-time PCR assays using the same mechanism - by hybridizing to their complementary DNA
targets. The products could be detected by adding the finished reaction to a microtiter well containing immobilized
molecular beacon probes and by reading the generated fluorescence. Alternatively, the closed-tube, real-time format
can be used. In this case, molecular beacons are added directly to the PCR mixture and hybridize to the newly
synthesized DNA in the course of the amplification reaction. Compared to TaqMan probes, molecular beacons are
advantageous in multiplex PCR assays because, in theory, they can determine more products simultaneously.
Q26. Phosphorothioate oligo as an antisense ODN.
As our understanding of molecular biology increases and we slowly gain insight on the molecular level of diseases,
the rational design of drugs becomes more feasible. In particular, molecules interacting on the level of proteins were
successfully designed and optimized for improved binding properties by bio-rational approaches. Synthetic
oligodeoxynucleotides (ODNs) have been proposed by Zamecnik and Stephenson as a new class of potential therapeutics
that can interact in a rational way with the messenger RNA (mRNA) of a disease related protein and thereby
specifically inhibit its synthesis (Figure).
The fascinating aspect of this approach, which is referred to as the antisense strategy, is the well-understood rules
of Watson-Crick base pairing by which single stranded nucleic acids interact with complementary oligonucleotides
forming a double helix. Every protein is synthesized by the principal mechanism of molecular biology: a gene
(double stranded DNA) carrying the genetic information for a particular protein is transcribed to single stranded
mRNA as intermediate carrier of information, which then serves as a template for the protein synthesis (translation)
by ribosomes. Therefore the antisense strategy is not limited to certain types of diseases but should be widely
applicable. Naturally occurring oligonucleotides (DNA and RNA) do not meet the criteria for potential antisense
drug candidates, and different chemical modifications were proposed to overcome the existing hurdles, which are
cellular uptake, resistance to nuclease and binding affinity toward mRNA. First, the oligodeoxynucleotides (ODNs)
must be able to cross the cellular membrane to reach the cytoplasm or nucleus. Once inside the cell the ODNs must
be resistant to degradation caused by nucleases. Finally, the ODNs must be able to bind specifically and with
high-affinity to the mRNA target in order to inhibit expression of the disease causing gene. The observed
activity of ODNs in tissue culture was earlier thought to imply that these theoretical barriers were not
a concern. However, analysis of the data from recent articles indicates that these theoretical barriers
are indeed real.
The focus of the recent researches is on the chemical approaches to improve the properties of ODNs, mainly concentrating
on the increase of nuclease resistance and mRNA binding affinity. The mRNA binding affinity is of high importance for
the antisense strategy and usually is reflected by “melting temperatures” (Tm's). Due to the p-p interaction of
stacked bases in duplex nucleic acids, the UV absorption at 260 nm is quenched. Upon heating, the annealed strands
denature and become single stranded, resulting in an increased absorption at 260 nm. Ideally, an S-shaped curve
is obtained from which, assuming a two state model, the thermodynamic parameters can be calculated. The midpoint
of this absorption vs. temperature profile is defined as Tm, reflecting the temperature at which equal fractions
of oligonucleotides are paired and single stranded under equilibrium. For the oligonucleotide sequences and lengths,
the following rule of thumb can be applied: an increase of Tm by 3-5°C(ΔTm) corresponds roughly to a 10-fold
increase of the association constant.
Modifications of the oligonucleotides have been made mainly on the phosphodiester backbone. As the hydrolytic cleavage
of the phosphodiester backbone is the main cause for the rapid degradation of oligonucleotides by nucleases, replacement
by other moieties has been one of the major strategies to improve stability. In addition, backbone modifications cam
influence other properties of oligonucleotides like RNA binding affinity or behavior for cellular uptake.
A first generation of backbone modifications retained the phosphorus atom as in the phosphorothioates. This derivative
was shown to possess an increased resistance toward nucleases. Therefore, the corresponding oligonucleotides could be
used for in vitro and in vivo experiments, but unfortunately they all displayed a lower binding affinity to the RNA
target. Nevertheless, phosphorothioates can be considered as oligonucleotide analogs of the first generation and were
shown to be biologically active against different molecular targets. This beneficial behavior is mainly due to
biological and chemical stability, favorable pharmacokinetic properties including cellular uptake, and RNase H
promoted mRNA cleavage observed for this class of compounds.
Phosphorothioates can be synthesized mainly by two different approaches: by the action of a solution of elemental sulfur
(S8) in carbon disulfide, or by the more recent method of using Beaucage's reagent (3H-1,2-Benzodithiol-1-one 1,1-dioxide
) sulfurizing phosphite triesters. The latter method avoids the problems of elemental sulfur's insolubility in most
organic solvents and the toxicity of carbon disulfide. Beaucage's reagent also yields phosphorothioates with higher
purity.
Phosphorothioate synthesis yields two isomers (R and S) at each incorporation site resulting in 2n-1 diastereoisomers
for an oligo of length n. Initially, there was much concern that this isomer mix would give unpredictable results.
Fortunately, however, the isomer mix in any standard S-oligo preparation does not appear to affect the biological
activity of the oligonucleotides.
Experiment
Q27. How do I make double stranded DNA?
To make double stranded DNA oligo from single stranded oligos, a simple yet effective annealing process is used.
However, it is imperative to be very cautious to remove undesired single stranded material.
• Make highly concentrated (1-10 OD260 units/100 mL) solutions of the oligos in STE buffer. (10 mM Tris-HCl pH 8.0,
50 nM NaCl, 1 mM EDTA)
• Mix the two equimolar solutions of the single stranded oligos.
• Heat to 94°C and slowly cool to room temperature.
• To ensure that complicated hairpin oligos are not formed, cool down very slowly.
• The final product, double stranded DNA, is stable and it is recommended to store at 4°C or in the freezer.